Detect-seq
A chemical labeling and biotin pulldown approach for the unbiased, genome-wide off-target evaluation of programmable cytosine base editors
Update log
- 2021-07-01 first commit. By MENG Haowei
- 2022-08-19 update version. Codes and algorithm design By MENG Haowei. Website, repository and document management By ZHAO Huanan.
What is Detect-seq?
Detect-seq aims to find all reliable CBE/DdCBE induced off-target editings on the whole genome.
This set of experiments and analytical procedures were reported by the YiLab @ Peking University in 2021.
Find more at www.detect-seq.com
What is Detect-seq tool?
Detect-seq tool can help find off-target editings, sgRNA alignments, and performing visualization of results. One can carry on Detect-seq experiments and then analyze Detect-seq data with the Detect-seq tool.
Citation
Please cite our publications if Detect-seq is used in your research.
# CBE & Detect-seq
1. Lei, Z., Meng, H., Lv, Z., Liu, M., Zhao, H., Wu, H., ... & Yi, C. (2021). Detect-seq reveals out-of-protospacer editing and target-strand editing by cytosine base editors. Nature Methods, 18(6), 643-651.
# DdCBE & Detect-seq
2. Lei, Z., Meng, H., Liu, L., Zhao, H., Rao, X., Yan, Y., ... & Yi, C. (2022). Mitochondrial base editor induces substantial nuclear off-target mutations. Nature, 606(7915), 804-811.
Environment
git clone https://github.com/menghaowei/Detect-seq.git
cd Detect-seq
# this conda env was tested on linux
conda env create -f conda-env.yaml
conda activate DetectSeq
configuration about Hisat-3n
The best practice of Detect-seq analysis
In order to comprehensively present the Detect-seq analysis workflow, here we selected two representative Detect-seq datasets from samples of CRISPR-Cas based CBE (BE4max) and CRISPR-Cas free mitochondrial base editor (DdCBE) for bioinformatics analysis demonstration. The relative GEO accessions are GSE151265 and GSE173859. More specifically, one can download the Detect-seq data of BE4max with VEGFA_site2 sgRNA and a matched mCherry control sample under the accession GSM6416152 and GSM6416156; one can download the Detect-seq data of DdCBE-L1397N-ND6 and a matched GFP control sample under the accession GSM5281973 and GSM5281964. The GEO download instructions can be found at https://www.ncbi.nlm.nih.gov/geo/info/download.html And we rename the raw sequencing FASTQ file as follows.
# BE4max VEGFA_site2 [GSM6416152]
Detect-293T-BE4max-VEGFA-All-PD_R1.fastq.gz
Detect-293T-BE4max-VEGFA-All-PD_R2.fastq.gz
# mCherry control [GSM6416156]
Detect-293T-BE4max-mCherry-PD_R1.fastq.gz
Detect-293T-BE4max-mCherry-PD_R2.fastq.gz
# DdCBE-L1397N-ND6 [GSM5281973]
Detect-293T-DdCBE-ND6-All-PD_R1.fastq.gz
Detect-293T-DdCBE-ND6-All-PD_R2.fastq.gz
# GFP control [GSM5281964]
Detect-293T-DdCBE-GFP-PD_R1.fastq.gz
Detect-293T-DdCBE-GFP-PD_R2.fastq.gz
Analysis Protocol
- 0. Download reference genome
- 1. Map the Detect-seq reads
- 2. Low mapping quality reads re-alignment
- 3. Duplication removal and reads cleaning of BAM files
- 4. Tracing tandem C-to-T signals and perform statistical test
- 5. Criteria selection and downstream analysis
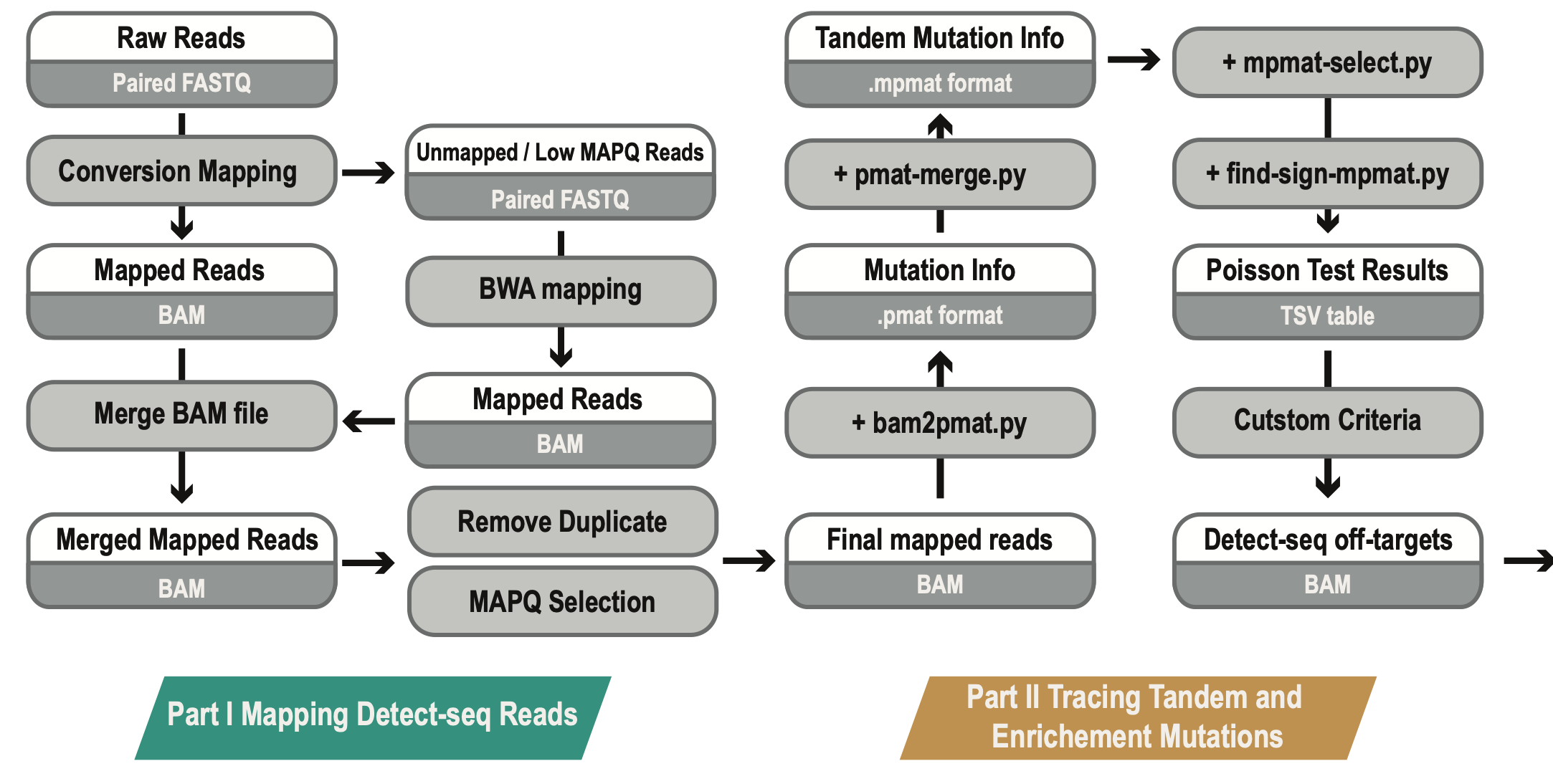
All Scripts and Codes can be found at /src/detect_seq
All Output Examples can be found at /data/example_results
We also offer a SnakeMake workflow at /snakemake/
0. Download reference genome
# Download reference genome
for index in {1..22} X Y M;
do
wget -c https://hgdownload.soe.ucsc.edu/goldenPath/hg38/chromosomes/chr${index}.fa.gz
done
# merge reference FASTA
fa_file=""
for index in {1..22} X Y M;
do
fa_file=${fa_file}" "chr${index}.fa.gz
done
cat $fa_file > hg38_only_chromosome.fa.gz
# decompression
gzip -d hg38_only_chromosome.fa.gz &
# build FASTA index
samtools faidx hg38_only_chromosome.fa
# build BWA MEM index
mkdir bwa_hg38
cd bwa_hg38
cp ../hg38_only_chromosome.fa* ./
bwa index hg38_only_chromosome.fa hg38_only_chromosome.fa > bwa_build.log 2>&1 &
# build HISAT3N index
mkdir hisat3n_hg38_CT
cp ./hg38_only_chromosome.fa* ./hisat3n_hg38_CT
hisat-3n-build --base-change C,T hg38_only_chromosome.fa hg38_only_chromosome.fa > hisat3n_hg38_CT_index.log 2>&1 &
1. Map the Detect-seq reads
Map the Detect-seq sequencing reads to the reference genome by HISAT-3N, which uses a base conversion alignment strategy.
# remove sequencing adapter
mkdir fix.fastq
for sample in 293T-BE4max-mCherry-PD 293T-BE4max-VEGFA-All-PD 293T-DdCBE-GFP-PD 293T-DdCBE-ND6-All-PD
do
in_fq_R1=raw.fastq/Detect-${sample}_R1.fastq.gz
in_fq_R2=raw.fastq/Detect-${sample}_R2.fastq.gz
out_fq_R1=fix.fastq/Detect-${sample}_R1_cutadapt.fq.gz
out_fq_R2=fix.fastq/Detect-${sample}_R2_cutadapt.fq.gz
log=fix.fastq/Detect-${sample}_cutadapt.log
cutadapt -j 20 --times 1 -e 0.1 -O 3 --quality-cutoff 25 -m 55 -a AGATCGGAAGAGCACACGT -A AGATCGGAAGAGCGTCGTG -o ${out_fq_R1} -p ${out_fq_R2} ${in_fq_R1} ${in_fq_R2} > ${log} 2>&1 &
done
# HISAT-3N mapping
mkdir bam.hisat3n
for sample in 293T-BE4max-mCherry-PD 293T-BE4max-VEGFA-All-PD 293T-DdCBE-GFP-PD 293T-DdCBE-ND6-All-PD
do
in_fq_R1=fix.fastq/Detect-${sample}_R1_cutadapt.fq.gz
in_fq_R2=fix.fastq/Detect-${sample}_R2_cutadapt.fq.gz
out_bam=bam.hisat3n/${sample}_hisat3n_hg38.bam
ummapped_fq=bam.hisat3n/${sample}_hisat3n_hg38_unmapped.fq.gz
log=bam.hisat3n/${sample}_hisat3n.log
ref_idx=reference/hisat3n_hg38_CT/hg38_only_chromosome.fa
hisat-3n -x ${ref_idx} -1 ${in_fq_R1} -2 ${in_fq_R2} -p 20 --sensitive --base-change C,T --unique-only --repeat-limit 1000 --no-spliced-alignment -X 700 --un-conc-gz ${ummapped_fq} --summary-file ${log} --rg-id ${sample} --rg "PL:ILLUMINA" --rg "ID:"${sample} --rg "SM:"${sample} | samtools view -hb > ${out_bam} &
done
2. Low mapping quality reads re-alignment
Merge the low mapping quality reads or unmapped reads and subject them to a re-alignment step. In this step, we utilize a non-conversion aligner like BWA MEM to re-align those reads to the reference genome.
# select low mapping quality BAM
for sample in 293T-BE4max-mCherry-PD 293T-BE4max-VEGFA-All-PD 293T-DdCBE-GFP-PD 293T-DdCBE-ND6-All-PD
do
in_bam=bam.hisat3n/${sample}_hisat3n_hg38.bam
out_bam=bam.hisat3n/${sample}_hisat3n_hg38.MAPQ20.LowerMAPQ20.bam
samtools view -h -@ 4 ${in_bam} | awk '$1~"@" || $5 <= 20 {print $0}' | samtools view -@ 4 -hb > ${out_bam} &
done
# BAM sort by reads name
for sample in 293T-BE4max-mCherry-PD 293T-BE4max-VEGFA-All-PD 293T-DdCBE-GFP-PD 293T-DdCBE-ND6-All-PD
do
in_bam=bam.hisat3n/${sample}_hisat3n_hg38.MAPQ20.LowerMAPQ20.bam
out_bam=bam.hisat3n/${sample}_hisat3n_hg38.MAPQ20.LowerMAPQ20.SortName.bam
temp_file=bam.hisat3n/${sample}_hisat3n_hg38.MAPQ20.LowerMAPQ20.SortName.bam.temp
samtools sort -O BAM -o ${out_bam} -T ${temp_file} -@ 15 -m 2G -n ${in_bam} &
done
# fetch low mapping quality reads from BAM file
for sample in 293T-BE4max-mCherry-PD 293T-BE4max-VEGFA-All-PD 293T-DdCBE-GFP-PD 293T-DdCBE-ND6-All-PD
do
in_bam=bam.hisat3n/${sample}_hisat3n_hg38.MAPQ20.LowerMAPQ20.SortName.bam
ref_genome_fa=reference/hisat3n_hg38_CT/hg38_only_chromosome.fa
out_fq_R1=bam.hisat3n/${sample}_hisat3n_hg38.LowerMAPQ20_R1.fq.gz
out_fq_R2=bam.hisat3n/${sample}_hisat3n_hg38.LowerMAPQ20_R2.fq.gz
samtools fastq -@ 15 -0 /dev/null -s /dev/null -n -F 0x900 -1 ${out_fq_R1} -2 ${out_fq_R2} --reference ${ref_genome_fa} ${in_bam} &
done
# merge unmapped reads and low mapping quality reads
for sample in 293T-BE4max-mCherry-PD 293T-BE4max-VEGFA-All-PD 293T-DdCBE-GFP-PD 293T-DdCBE-ND6-All-PD
do
low_fq_R1=bam.hisat3n/${sample}_hisat3n_hg38.LowerMAPQ20_R1.fq.gz
low_fq_R2=bam.hisat3n/${sample}_hisat3n_hg38.LowerMAPQ20_R2.fq.gz
unmapped_fq_R1=bam.hisat3n/${sample}_hisat3n_hg38_unmapped.fq.1.gz
unmapped_fq_R2=bam.hisat3n/${sample}_hisat3n_hg38_unmapped.fq.2.gz
out_fq_R1=bam.hisat3n/${sample}_R1_unmapped_and_LowerMAPQ20.fq.gz
out_fq_R2=bam.hisat3n/${sample}_R2_unmapped_and_LowerMAPQ20.fq.gz
cat ${low_fq_R1} ${unmapped_fq_R1} > ${out_fq_R1} &
cat ${low_fq_R2} ${unmapped_fq_R2} > ${out_fq_R2} &
done
# re-alignment with BWA MEM
for sample in 293T-BE4max-mCherry-PD 293T-BE4max-VEGFA-All-PD 293T-DdCBE-GFP-PD 293T-DdCBE-ND6-All-PD
do
in_fq_R1=bam.hisat3n/${sample}_R1_unmapped_and_LowerMAPQ20.fq.gz
in_fq_R2=bam.hisat3n/${sample}_R2_unmapped_and_LowerMAPQ20.fq.gz
bwa_index=reference/bwa_hg38/hg38_only_chromosome.fa
out_bam=bam.hisat3n/${sample}_bwa_hg38_realign.bam
bwa_log=bam.hisat3n/${sample}_bwa_hg38_realign.log
bwa mem ${bwa_index}${in_fq_R1} {in_fq_R2} -t 20 -M -R '@RG\tID:'${sample}'\tPL:ILLUMINA\tSM:'${sample} 2>${bwa_log} | samtools view -h -b -q 20 -f 3 -F 256 > ${out_bam} &
done
3. Duplication removal and reads cleaning of BAM files
Merge the HISAT-3N alignment BAM file and the BWA MEM alignment BAM file together, and then remove the duplication reads, non-concordant reads, clipped mapping reads and the secondary alignment reads. Only reads with a mapping quality larger than 20 are retained for subsequent analysis.
# merge HISAT-3n BAM and BWA MEM BAM
for sample in 293T-BE4max-mCherry-PD 293T-BE4max-VEGFA-All-PD 293T-DdCBE-GFP-PD 293T-DdCBE-ND6-All-PD
do
in_bam_bwa=bam.hisat3n/${sample}_bwa_hg38_realign.bam
in_bam_hisat3n=bam.hisat3n/${sample}_hisat3n_hg38.bam
out_bam=bam.hisat3n/${sample}_hg38_merge.MAPQ20.bam
samtools cat -o ${out_bam} ${in_bam_hisat3n} ${in_bam_bwa} &
done
# sort BAM by genome coordinate
for sample in 293T-BE4max-mCherry-PD 293T-BE4max-VEGFA-All-PD 293T-DdCBE-GFP-PD 293T-DdCBE-ND6-All-PD
do
in_bam=bam.hisat3n/${sample}_hg38_merge.MAPQ20.bam
out_bam=bam.hisat3n/${sample}_hg38_merge_sort.MAPQ20.bam
temp_file=bam.hisat3n/${sample}_hg38_merge_sort.MAPQ20.bam.temp
samtools sort -O BAM -o ${out_bam} -T ${temp_file} -@ 15 -m 2G ${in_bam} &
done
# remove duplication
for sample in 293T-BE4max-mCherry-PD 293T-BE4max-VEGFA-All-PD 293T-DdCBE-GFP-PD 293T-DdCBE-ND6-All-PD
do
in_bam=bam.hisat3n/${sample}_hg38_merge_sort.MAPQ20.bam
out_log=bam.hisat3n/${sample}_hg38_merge_sort_rmdup.log
out_bam=bam.hisat3n/${sample}_hg38_merge_sort_rmdup.MAPQ20.WithClip.bam
out_matrix=bam.hisat3n/${sample}_hg38_merge_sort_rmdup.matrix
java -Xms50g -Xmx50g -XX:ParallelGCThreads=20 -jar picard.jar MarkDuplicates I=${in_bam} O=${out_bam} M=${out_matrix} ASO=coordinate REMOVE_DUPLICATES=true 2> ${out_log} &
done
# filter clip, non-concordant reads, low MAPQ reads and secondary alignment
for sample in 293T-BE4max-mCherry-PD 293T-BE4max-VEGFA-All-PD 293T-DdCBE-GFP-PD 293T-DdCBE-ND6-All-PD
do
in_bam=bam.hisat3n/${sample}_hg38_merge_sort_rmdup.MAPQ20.WithClip.bam
out_bam=bam.hisat3n/${sample}_hg38_merge_sort_rmdup.MAPQ20.bam
ref_genome_fa=reference/hisat3n_hg38_CT/hg38_only_chromosome.fa
samtools view -@ 4 -h ${in_bam} -q 20 -f 3 -F 256 | samclip --ref ${ref_genome_fa} --max 3 --progress 0 | awk 'function abs(v) {return v < 0 ? -v : v} $1~"@" || ($7 == "=" && abs($9) <= 2500 ) {print $0}' | samtools view -@ 4 -hb > ${out_bam} &
done
# build BAM index
for sample in 293T-BE4max-mCherry-PD 293T-BE4max-VEGFA-All-PD 293T-DdCBE-GFP-PD 293T-DdCBE-ND6-All-PD
do
in_bam=bam.hisat3n/${sample}_hg38_merge_sort_rmdup.MAPQ20.bam
out_bam_index=bam.hisat3n/${sample}_hg38_merge_sort_rmdup.MAPQ20.bam.bai
samtools index -@ 10 ${in_bam} ${out_bam_index} &
done
4. Tracing tandem C-to-T signals and perform statistical test
Catch contiguous tandem mutation information from BAM files (i.e., tandem C-to-T mutation signals for the Watson strand and tandem G-to-A mutation signals for the Crick strand). Then perform a comparison for those tandem mutation signals between control sample and Detect-seq treatment sample by the Poisson statistical test. After this step, one can obtain tables containing information of tandem mutation signals (a mpmat file shown in the code part) and matched Poisson test results (a TSV table shown in the code part).
# convert BAM to pmat format
mkdir pmat_and_mpmat
for sample in 293T-BE4max-VEGFA-All-PD 293T-DdCBE-ND6-All-PD
do
in_bam=bam.hisat3n/${sample}_hg38_merge_sort_rmdup.MAPQ20.bam
ref_genome_fa=reference/hisat3n_hg38_CT/hg38_only_chromosome.fa
out_pmat=pmat_and_mpmat/${sample}_hg38_merge_sort_rmdup.MAPQ20.pmat
out_log=pmat_and_mpmat/${sample}_hg38_merge_sort_rmdup.MAPQ20.bam2pmat.log
python bam2pmat.py -i ${in_bam} -r ${ref_genome_fa} -o ${out_pmat} -p 20 --out_format pmat --bed_like_format True --mut_type ALL --block_size 100000 --cover_num_cutoff 0 --mut_num_cutoff 0 --mut_ratio_cutoff 0 --keep_temp_file False --out_header False > ${out_log} 2>&1 &
done
# select
# tracing tandem mutation signals
for sample in 293T-BE4max-VEGFA-All-PD 293T-DdCBE-ND6-All-PD
do
ref_genome_fa=reference/hisat3n_hg38_CT/hg38_only_chromosome.fa
in_pmat=pmat_and_mpmat/${sample}_hg38_merge_sort_rmdup.MAPQ20.pmat
out_C_pmat=pmat_and_mpmat/${sample}_hg38_merge_sort_rmdup.MAPQ20_C.pmat
out_G_pmat=pmat_and_mpmat/${sample}_hg38_merge_sort_rmdup.MAPQ20_G.pmat
out_CT_mpmat=pmat_and_mpmat/${sample}_hg38.MAPQ20.merge_d50_D100.CT.mpmat
out_GA_mpmat=pmat_and_mpmat/${sample}_hg38.MAPQ20.merge_d50_D100.GA.mpmat
out_CT_log=pmat_and_mpmat/${sample}_hg38.MAPQ20.merge_d50_D100.CT.mpmat.log
out_GA_log=pmat_and_mpmat/${sample}_hg38.MAPQ20.merge_d50_D100.GA.mpmat.log
# CT on the Watson strand
awk '$10 == "C" {print $0}' ${in_pmat} > ${out_C_pmat} &
python pmat-merge.py -i ${out_C_pmat} -f C -t T -r ${ref_genome_fa} -d 50 -D 100 --NoMutNumCutoff 2 --OmitTandemNumCutoff 2 -o ${out_CT_mpmat} --SNP snv_info/293T-snv_info.list1.bed,snv_info/293T-snv_info.list2.vcf 2> ${out_CT_log} &
# CT on the Crick strand
awk '$10 == "G" {print $0}' ${in_pmat} > ${out_G_pmat} &
python pmat-merge.py -i ${out_G_pmat} -f G -t A -r ${ref_genome_fa} -d 50 -D 100 --NoMutNumCutoff 2 --OmitTandemNumCutoff 2 -o ${out_GA_mpmat} --SNP snv_info/293T-snv_info.list1.bed,snv_info/293T-snv_info.list2.vcf 2> ${out_GA_log} &
done
# merge tandem mutation signals and sort
for sample in 293T-BE4max-VEGFA-All-PD 293T-DdCBE-ND6-All-PD
do
in_CT_mpmat=pmat_and_mpmat/${sample}_hg38.MAPQ20.merge_d50_D100.CT.mpmat
in_GA_mpmat=pmat_and_mpmat/${sample}_hg38.MAPQ20.merge_d50_D100.GA.mpmat
ref_genome_fa_index=reference/hisat3n_hg38_CT/hg38_only_chromosome.fa.fai
out_CT_mpmat=pmat_and_mpmat/${sample}_hg38.MAPQ20.merge_d50_D100.CT.select.mpmat
out_GA_mpmat=pmat_and_mpmat/${sample}_hg38.MAPQ20.merge_d50_D100.GA.select.mpmat
out_merge_mpmat=pmat_and_mpmat/${sample}_hg38.MAPQ20.merge_d50_D100.merge.select.mpmat
out_merge_sort_mpmat=pmat_and_mpmat/${sample}_hg38.MAPQ20.merge.select.sort.mpmat
out_rm_chr_mpmat=pmat_and_mpmat/${sample}_hg38.MAPQ20.merge.select.sort.RmChrYChrM.mpmat
# select CT
python mpmat-select.py -i {in_CT_mpmat} -o ${out_CT_mpmat} -f C -t T -m 4 -c 6 -r 0.01 --RegionPassNum 1 --RegionToleranceNum 10 --RegionMutNum 2 --InHeader True --OutHeader False
# select GA
python mpmat-select.py -i {in_GA_mpmat} -o ${out_GA_mpmat} -f G -t A -m 4 -c 6 -r 0.01 --RegionPassNum 1 --RegionToleranceNum 10 --RegionMutNum 2 --InHeader True --OutHeader False
# merge CT singal on the Watson strand and the Crick strand
cat ${out_CT_mpmat} ${out_GA_mpmat} > ${out_merge_mpmat}
# sort by the genome coordinate
bedtools sort -i ${out_merge_mpmat} -g ${ref_genome_fa_index} | uniq > ${out_merge_sort_mpmat}
# remove chrY and chrM
cat ${out_merge_sort_mpmat} | grep -v chrY | grep -v chrM > ${out_rm_chr_mpmat}
done
# run the Poisson enrichment test
ref_genome_fa=reference/hisat3n_hg38_CT/hg38_only_chromosome.fa
# for BE4max sample
python find-significant-mpmat.py -p 25 \
-i pmat_and_mpmat/293T-BE4max-VEGFA-All-PD_hg38.MAPQ20.merge.select.sort.RmChrYChrM.mpmat \
-o poisson_res/293T-BE4max-VEGFA-All-PD_vs_ctrl_hg38.select.pvalue_table \
-c bam.hisat3n/293T-BE4max-mCherry-PD_hg38_merge_sort_rmdup.MAPQ20.bam \
-t bam.hisat3n/293T-BE4max-VEGFA-All-PD_hg38_merge_sort_rmdup.MAPQ20.bam \
-r ${ref_genome_fa} \
--query_mutation_type CT,GA \
--mpmat_filter_info_col_index -1 \
--mpmat_block_info_col_index -1 \
--region_block_mut_num_cutoff 2 \
--query_mut_min_cutoff 2 \
--query_mut_max_cutoff 16 \
--total_mut_max_cutoff 16 \
--other_mut_max_cutoff 6 \
--seq_reads_length 150 \
--lambda_method ctrl_max \
--poisson_method mutation \
2> poisson_res/293T-BE4max-VEGFA-All-PD_vs_ctrl_hg38_possion_test.log &
# for DdCBE sample
python find-significant-mpmat.py -p 25 \
-i pmat_and_mpmat/293T-DdCBE-ND6-All-PD_hg38.MAPQ20.merge.select.sort.RmChrYChrM.mpmat \
-o poisson_res/293T-DdCBE-ND6-All-PD_vs_ctrl_hg38.select.pvalue_table \
-c bam.hisat3n/293T-DdCBE-GFP-PD_hg38_merge_sort_rmdup.MAPQ20.bam \
-t bam.hisat3n/293T-DdCBE-ND6-All-PD_hg38_merge_sort_rmdup.MAPQ20.bam \
-r ${ref_genome_fa} \
--query_mutation_type CT,GA \
--mpmat_filter_info_col_index -1 \
--mpmat_block_info_col_index -1 \
--region_block_mut_num_cutoff 2 \
--query_mut_min_cutoff 2 \
--query_mut_max_cutoff 16 \
--total_mut_max_cutoff 16 \
--other_mut_max_cutoff 6 \
--seq_reads_length 150 \
--lambda_method ctrl_max \
--poisson_method mutation \
2> poisson_res/293T-DdCBE-ND6-All-PD_vs_ctrl_hg38_possion_test.log &
doc for the output pvalue_table
An output example for the enrichment significance test results.
header explaination
chr_name: str, chromosome name of tested region, value like ‘chr1’, ‘chr2’ …region_start: int, the start coordinate of the tested region, and the coordinate index is based on a 1-based scale.region_end: int, the end coordinate of the tested region, and the coordinate index is based on a 1-based scale.mpmat_index: str, a formatted string, which can be used as a key to index the whole table.region_site_num: int, number of sites (C or G) in the tested regionregion_block_site_num: int, number of sites that present a mutation signal in the control sample (C-to-T or G-to-A). The blocked sites are omitted in the enrichment test step.region_mut_site_num: int, number of sites with mutated signals in the treatment sample. Note, the blocked sites are not considered.region_site_index: str list, split by comma, list length is the same as, and each item in this list is the site coordinate of the genome. region_block_state: str list, split by -, list length is the same as, “B” means site is blocked, and "N" means site is not blocked. region_highest_site_index: str, coordinate of site with the highest Detect-seq signal.region_highest_site_mut_num: int, count of sequencing reads with tandem mutation info for the site with the highest Detect-seq signal.region_highest_site_cover_num: int, total count of sequencing reads for the site with the highest Detect-seq signal.region_highest_site_mut_ratio: float, range 0~1, mutation ratio, which equals/ ctrl_count: int, total count of sequencing reads in the control sample; if a read overlaps with the tested region, it will be counted.treat_count: int, total count of sequencing reads in treat sample.ctrl_mut_count: int, count of sequencing reads with C-to-T / G-to-A mutation info in control sample.treat_mut_count: int, count of sequencing reads with C-to-T / G-to-A mutation info in treat sample.ctrl_count.nor:, float, normalized, the default value equals count per million. treat_count.nor:, float, normalized, the default value equals count per million. ctrl_mut_count.nor:, float, normalized, the default value equals count per million. treat_mut_count.nor:, float, normalized, the default value equals count per million. count_info: meaning less in this version.log2_FC: float, log2 fold-change, which equals log2(/ ) log2_FC_mut: float, log2 fold-change, which equals log2(/ ) test_state: str, “TestOK” means the Poisson enrichment test works well.p_value: float, the p-value from the Poisson enrichment test.FDR: float, adjusted p-value with BH methods.
chr_name region_start region_end mpmat_index region_site_num region_block_site_num region_mut_site_num region_site_index region_block_state region_highest_site_index region_highest_site_mut_num region_highest_site_cover_num region_highest_site_mut_ratio ctrl_count treat_count ctrl_mut_count treat_mut_count ctrl_count.norm treat_count.norm ctrl_mut_count.norm treat_mut_count.norm count_info log2_FC log2_FC_mut test_state p_value FDR
chr1 49272 49288 chr1_49272_49288 3 1 2 "chr1_49272_GA,chr1_49276_G.,chr1_49288_GA" B-N-N chr1_49288_GA 1 5 0.2 11 10 0 0 0.080257214 0.051644633 0 0 "0,1,2,3 10,1,0,0 9,1,0,0" -0.63601266 NA TestOK 0.5 0.511034942
chr1 49515 49529 chr1_49515_49529 3 0 3 "chr1_49515_GA,chr1_49521_G.,chr1_49529_GA" N-N-N chr1_49515_GA 1 3 0.333333333 9 12 1 1 0.065664993 0.061973559 0.00729611 0.005164463 "0,1,2,3 0,8,1,0 3,8,1,0" -0.083471637 -0.498509136 TestOK 0.5 0.511034942
chr1 73840 73842 chr1_73840_73842 3 1 2 "chr1_73840_CT,chr1_73841_CT,chr1_73842_CT" N-B-N chr1_73840_CT 1 16 0.0625 16 22 0 0 0.116737766 0.113618192 0 0 "0,1,2,3 13,3,0,0 20,2,0,0" -0.039077518 NA TestOK 0.5 0.511034942
chr1 99045 99092 chr1_99045_99092 10 0 10 "chr1_99045_CT,chr1_99046_CT,chr1_99055_CT,chr1_99059_CT,chr1_99061_CT,chr1_99069_CT,chr1_99072_CT,chr1_99083_CT,chr1_99088_CT,chr1_99092_CT" N-N-N-N-N-N-N-N-N-N chr1_99045_CT 1 6 0.166666667 18 14 9 7 0.131329987 0.072302486 0.065664993 0.036151243 "0,1,2,3,4,5,6,7,8,9,10 7,1,2,1,0,2,1,0,1,0,3 7,0,2,0,0,0,1,2,0,0,2" -0.861079216 -0.861079216 TestOK 0.687775284 0.699676496
chr1 144871 144900 chr1_144871_144900 4 0 4 "chr1_144871_GA,chr1_144873_GA,chr1_144875_GA,chr1_144900_GA" N-N-N-N chr1_144871_GA 2 10 0.2 7 15 1 3 0.051072773 0.077466949 0.00729611 0.01549339 "0,1,2,3,4 1,5,0,1,0 5,7,0,3,0" 0.601026537 1.086453364 TestOK 0.173669361 0.32172689
chr1 184167 184193 chr1_184167_184193 9 0 9 "chr1_184167_GA,chr1_184168_GA,chr1_184170_GA,chr1_184176_GA,chr1_184177_GA,chr1_184178_GA,chr1_184185_GA,chr1_184188_GA,chr1_184193_GA" N-N-N-N-N-N-N-N-N chr1_184167_GA 4 24 0.166666667 19 33 0 4 0.138626097 0.170427288 0 0.020657853 "0,1,2,3,4,5,6,7,8,9 15,4,0,0,0,0,0,0,0,0 27,2,2,0,0,1,0,0,0,1" 0.29795747 1.130606173 TestOK 0.01821861 0.094641139
chr1 191997 192025 chr1_191997_192025 3 1 2 "chr1_191997_GA,chr1_192015_G.,chr1_192025_GA" N-N-B chr1_191997_GA 1 14 0.071428571 7 16 0 0 0.051072773 0.082631412 0 0 "0,1,2,3 7,0,0,0 14,2,0,0" 0.694135942 NA TestOK 0.5 0.511034942
chr1 271391 271413 chr1_271391_271413 5 2 3 "chr1_271391_CT,chr1_271396_CT,chr1_271401_CT,chr1_271406_CT,chr1_271413_CT" N-N-S-S-N chr1_271391_CT 1 7 0.142857143 5 13 0 2 0.036480552 0.067138022 0 0.010328927 "0,1,2,3,4,5 4,1,0,0,0,0 5,6,1,1,0,0" 0.880002487 0.130606173 TestOK 0.094520046 0.209966519
...
5. Criteria selection and downstream analysis
5.1 Criteria for selection of significant Detect-seq signals
Select the significant Detect-seq signals according to the Poisson test results with a certain criterion. Note that an arbitrary criterion could introduce potential false positive or false negative issues. In fact, Detect-seq of BE4max and DdCBE samples show different mutation patterns, which require corresponding thresholds. Here, we recommend two criteria, one strict and another lenient, for users to select the significant Detect-seq signals of BE4max or DdCBE samples. The following codes can run in an R session.
# 1. BE4max
rm(list=ls())
library(tidyverse)
setwd(dir = "./detect_nat_protocol/")
#-------------------------------------------------------------------------->>>>>>>
# 1.1 find sign region
#-------------------------------------------------------------------------->>>>>>>
# load merge table
poisson_res = read_tsv(file = "./poisson_res/293T-BE4max-VEGFA-All-PD_vs_ctrl_hg38.select.pvalue_table")
# check column name
colnames(poisson_res)
#-------------------------------------------------------------------------->>>>>>>
# select significant Detect-seq signals
#-------------------------------------------------------------------------->>>>>>>
# strict selection
poisson_res.sign.strict = filter(
poisson_res,
FDR < 0.0001 ,
log2_FC_mut >= 2,
ctrl_mut_count <= 1,
treat_mut_count.norm * 100 >= 10,
treat_mut_count >= 20,
treat_mut_count / treat_count >= 0.15,
region_block_site_num <= 1,
region_highest_site_mut_ratio >= 0.35
)
# lenient selection
poisson_res.sign.lenient = filter(
poisson_res,
FDR < 0.01 ,
log2_FC_mut >= 2,
ctrl_mut_count <= 1,
treat_mut_count.norm * 100 >= 5,
treat_mut_count >= 10,
treat_mut_count / treat_count >= 0.15,
region_block_site_num <= 1,
region_highest_site_mut_ratio >= 0.30
)
# --------------------------------------------------------------------------------->>>>>
# get corresponding mpmat file
# --------------------------------------------------------------------------------->>>>>
# mpmat table column type
col_type_info = cols(
X1 = col_character(),
X2 = col_double(),
X3 = col_double(),
X4 = col_double(),
X5 = col_double(),
X6 = col_double(),
X7 = col_character(),
X8 = col_character(),
X9 = col_character(),
X10 = col_character(),
X11 = col_character(),
X12 = col_character(),
X13 = col_character()
)
mpmat_filename = "./pmat_and_mpmat/293T-BE4max-VEGFA-All-PD_hg38.MAPQ20.merge.select.sort.RmChrYChrM.mpmat"
mpmat_table = read_tsv(file = mpmat_filename, col_names = F, col_types = col_type_info)
colnames(mpmat_table)[1:3] = c("chrom", "start", "end")
mpmat_table.AddIdx <- mpmat_table %>% unite("mpmat_index", chrom:end, sep = "_", remove = F)
# strict mpmat select
mpmat_table.strict.select <- filter(
mpmat_table.AddIdx,
mpmat_index %in% poisson_res.sign.strict$mpmat_index
) %>% select(
-mpmat_index
)
write_tsv(mpmat_table.strict.select, file = "./final_list/293T-BE4max-VEGFA.StrictList.mpmat", col_names = F)
# lenient mpmat select
mpmat_table.lenient.select <- filter(
mpmat_table.AddIdx,
mpmat_index %in% poisson_res.sign.lenient$mpmat_index
) %>% select(
-mpmat_index
)
write_tsv(mpmat_table.lenient.select, file = "./final_list/293T-BE4max-VEGFA.LenientList.mpmat", col_names = F)
# 2. DdCBE
rm(list=ls())
library(tidyverse)
setwd(dir = "./detect_nat_protocol/")
#-------------------------------------------------------------------------->>>>>>>
# 2.1 find sign region
#-------------------------------------------------------------------------->>>>>>>
# load merge table
poisson_res = read_tsv(file = "./poisson_res/293T-DdCBE-ND6-All-PD_vs_ctrl_hg38.select.pvalue_table")
# check column name
colnames(poisson_res)
# strict selection
poisson_res.sign.strict = filter(
poisson_res,
FDR < 0.0001 ,
log2_FC_mut >= 2,
ctrl_mut_count <= 1,
treat_mut_count.norm * 100 >= 10,
treat_mut_count >= 15,
treat_mut_count / treat_count >= 0.15,
region_block_site_num <= 1,
region_highest_site_mut_ratio >= 0.25
)
# lenient selection
poisson_res.sign.lenient.part1 = filter(
poisson_res,
FDR < 0.01 ,
log2_FC_mut >= 2,
ctrl_mut_count <= 1,
treat_mut_count.norm * 100 >= 5,
treat_mut_count >= 15,
treat_mut_count / treat_count >= 0.15,
region_block_site_num <= 1,
region_highest_site_mut_ratio >= 0.15
)
#-------------------------------------------------------------------------->>>>>>>
# get corresponding mpmat file
#-------------------------------------------------------------------------->>>>>>>
# mpmat table column type
col_type_info = cols(
X1 = col_character(),
X2 = col_double(),
X3 = col_double(),
X4 = col_double(),
X5 = col_double(),
X6 = col_double(),
X7 = col_character(),
X8 = col_character(),
X9 = col_character(),
X10 = col_character(),
X11 = col_character(),
X12 = col_character(),
X13 = col_character()
)
mpmat_filename = "./pmat_and_mpmat/293T-DdCBE-ND6-All-PD_hg38.MAPQ20.merge.select.sort.RmChrYChrM.mpmat"
mpmat_table = read_tsv(file = mpmat_filename, col_names = F, col_types = col_type_info)
colnames(mpmat_table)[1:3] = c("chrom", "start", "end")
mpmat_table.AddIdx <- mpmat_table %>% unite("mpmat_index", chrom:end, sep = "_", remove = F)
# strict mpmat select
mpmat_table.strict.select <- filter(
mpmat_table.AddIdx,
mpmat_index %in% poisson_res.sign.strict.select$mpmat_index
) %>% select(
-mpmat_index
)
dim(mpmat_table.strict.select)
write_tsv(mpmat_table.strict.select, file = "./final_list/293T-DdCBE-ND6.StrictList.mpmat", col_names = F)
# lenient mpmat select
mpmat_table.lenient.select <- filter(
mpmat_table.AddIdx,
mpmat_index %in% poisson_res.sign.lenient.select$mpmat_index
) %>% select(
-mpmat_index
)
dim(mpmat_table.lenient.select)
write_tsv(mpmat_table.lenient.select, file = "./final_list/293T-DdCBE-ND6.LenientList.mpmat", col_names = F)
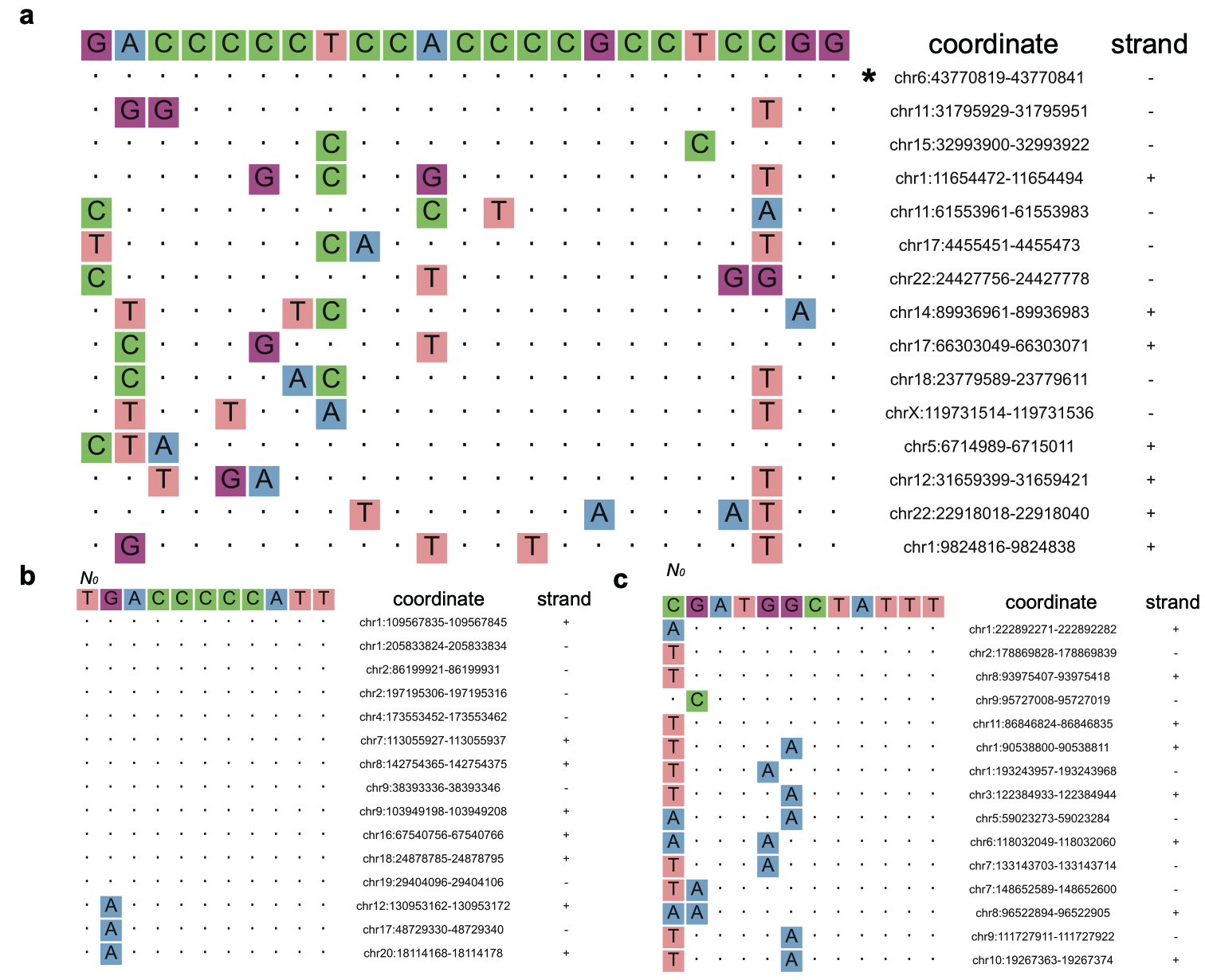
5.2 Query sequence alignment and results plot
After finishing the previous steps, one can obtain significantly enriched tandem mutation signals of Detect-seq, indicating the off-target regions caused by cytosine base editors. Run the following commands if one want to further find putative sgRNA binding site (pRBS) or putative TALE-array binding site (pTBS) among these regions.
# reference genome FASTA file
ref_genome_fa=reference/hisat3n_hg38_CT/hg38_only_chromosome.fa
# find VEGFA_site2 pRBS
python mpmat-to-art-sgRNA.py -i ./final_list/293T-BE4max-VEGFA.LenientList.mpmat -q GACCCCCTCCACCCCGCCTCNRG -r ${ref_genome_fa} -o ./final_list/293T-BE4max-VEGFA.LenientList.sgRNA_align.art \
-m region -e 50 --input_header False \
--mpmat_fwd_mut_type CT \
--mpmat_rev_mut_type GA \
--seed_index 15,20 \
--align_settings 5,-4,-24,-8 \
--PAM_type_penalty 0,8,12 \
--dna_bulge_penalty 24,8 \
--rna_bulge_penalty 24,8 \
--dna_bulge_cmp_weight 1,24 \
--rna_bulge_cmp_weight 1,24 \
--mismatch_cmp_weight 10,2 \
--dist_to_signal_penalty_k 0,0,0,0,0,0 \
--dist_to_signal_penalty_offset 12,0,0,0,0,12 &
# find ND6 left pTBS
python mpmat-to-art-TALE.py -r ${ref_genome_fa} -i ./final_list/293T-DdCBE-ND6.LenientList.mpmat -q TGACCCCCATT -m region -e 50 --input_filetype mpmat --input_header False -o ./final_list/293T-DdCBE-ND6.LenientList.TALE_align.Left.art &
# find ND6 right pTBS
python mpmat-to-art-TALE.py -r ${ref_genome_fa} -i ./final_list/293T-DdCBE-ND6.LenientList.mpmat -q CGATGGCTATTT -m region -e 50 --input_filetype mpmat --input_header False -o ./final_list/293T-DdCBE-ND6.LenientList.TALE_align.Right.art &
# plot VEGFA pRBS results
python plot-art.py \
-i ./final_list/293T-BE4max-VEGFA.LenientList.sgRNA_align.art \
-o ./out_image/293T-BE4max-VEGFA.LenientList.sgRNA_align.pdf \
--align_seq GACCCCCTCCACCCCGCCTCCGG \
-k align_total_match,align_total_mismatch,align_total_gap,seed_mismatch -r False,True,True,True \
-a align_coordinate,align_strand,align_total_mismatch,seed_mismatch,region_index &
# plot ND6 left pTBS results
python plot-art.py \
-i ./final_list/293T-DdCBE-ND6.LenientList.TALE_align.Left.art \
-o ./out_image/293T-DdCBE-ND6.LenientList.TALE_align.Left.pdf \
--align_seq TGACCCCCATT \
-k align_total_mismatch,align_degen_total_mismatch,align_total_gap -r True,True,True \
-a align_coordinate,align_strand,align_total_mismatch,align_degen_total_mismatch,region_index &
# plot ND6 right pTBS results
python plot-art.py \
-i ./final_list/293T-DdCBE-ND6.LenientList.TALE_align.Right.art \
-o ./out_image/293T-DdCBE-ND6.LenientList.TALE_align.Right.pdf \
--align_seq CGATGGCTATTT \
-k align_total_mismatch,align_degen_total_mismatch,align_total_gap -r True,True,True \
-a align_coordinate,align_strand,align_total_mismatch,align_degen_total_mismatch,region_index &